

背景及概述[1][2]
2,6-二溴可由对硝基溴代得到,可用于制备2,6-二溴溴苄。
制备[1]

1,3-二溴-2-甲基-5-硝基:向1-甲基-4-硝基(30.0g,218.8mmol)的CHCl3溶液(120mL)中加入铁粉(3.6g,64.5mmol)用机械搅拌。然后缓慢加入溴(124.8g,40mL,780.9mmol),同时将温度升至40℃。加入溴后,将混合物加热回流48小时。冷却后,将溶液用饱和Na2SO3溶液,饱和Na2CO3溶液,盐水洗涤,并经无水Na2SO4干燥。除去溶剂后,残余物用MeOH重结晶,得到26.5g标题化合物,为黄色晶体。通过二氧化硅柱色谱法获得另外的12.3g标题化合物。总收率:60%。 1,3-二溴-2-甲基-5-硝基,产率12.3g,60%。 1H NMR(400MHz,CDCl3)δ2.67(s,3H),8.38(s,2H)。
5-(3,5-二溴-4-甲基基)胺:将1,3-二溴-2-甲基-5-硝基(11.3g,38.3mmol)溶于THF / EtOH(100mL / 100mL)中,然后加入SnCl2 2H2O(43.2g,191.6mmol)。将混合物在室温下搅拌3小时。除去溶剂后,加入NaOH溶液(25g / 200mL),搅拌混合物1.5小时。将溶液用EtOAc(200mL×2)萃取并经无水Na2SO4干燥。除去EtOAc后,加入CH2Cl2,然后加入浓HCl(7mL),形成盐酸盐,通过过滤收集。该固体无需进一步纯化即可用于后续反应。(3,5-二溴-4-甲基基)胺。1 H NMR(400MHz,D2O)δ2.43(s,3H),3.61(br,2H),6.86(s,2H)。
1,3-二溴-2-甲基:将(3,5-二溴-4-甲基基)胺溶于水(80mL)和浓HCl(7.5mL)中的溶液搅拌20分钟,然后冷却混合物在0-5℃下加入NaNO2(3.4g / 40mL H2O)溶液。将反应混合物在0-5℃下搅拌2小时,然后将悬浮液加入到次磷酸(50%,27.9g)的溶液中,并将混合物冷却至0℃。将混合物在室温下搅拌过夜。然后用CH2Cl2(100mL×2)萃取。将有机层用盐水(30mL)洗涤并经Na2SO4干燥。在二氧化硅柱色谱法(用石油醚洗脱)后,得到3.57g产物,为无色液体。 1,3-二溴-2-甲基,产量3.57g。 1H NMR(400MHz,CDCl3)δ2.57(s,3H),6.89(t,J = 8.0Hz,1H),7.50(d,J = 8.0Hz,2H)。
应用[2]
2,6-二溴溴苄是一种重要的医药中间体,白三烯和前列腺素是炎症的重要介质,可以分别促进不同途径的炎症的发展,专利WO2006128142A2中以2,6-二溴溴苄为原料合成的胞质磷脂酶(cPLA2)可作为白三烯和前列腺素活性的化学抑制剂,达到治疗炎症的效果;Adri等以2,6-二溴溴苄为原料合成的罗平尼咯是一种用于治疗帕金森综合症的药物;Thomas等以2,6-二溴溴苄为原料合成的四氢喹啉化合物是一种新型胆固醇酯转运蛋白抑制剂。
2,6-二溴溴苄的合成是以2,6-二溴为原料,经溴代反应制得。该反应以N-溴代丁二酰亚胺作为溴化试剂,过氧化甲酰为引发剂,四氯化碳为溶剂,收率89.0%。该法虽然收率较高,但存在N-溴代丁二酰亚胺价格较贵、溶剂毒性大,过氧化甲酰引入杂质等问题。
CN201110000564.1提供了一种操作简单、经济的2,6-二溴溴苄的制备方法。该法采用氢溴酸与双氧水代替传统的溴化试剂N-溴代丁二酰亚胺;用光照代替引发剂过氧化甲酰;用无毒或较低毒性的溶剂代替高毒性的四氯化碳。
氢溴酸与双氧水反应产生溴素,并在光引发下生成的溴自由基,夺取甲基氢生成2,6-二溴苄基自由基,后与溴素反应生成2,6-二溴溴苄以及溴自由基,后者继续参与下一轮自由基取代反应。传统溴素取代法,生成一分子溴代产物的同时,也生成一分子强腐蚀性的溴化氢,原料利用率低,且不利于环保。本发明中溴化氢始终处于反应循环,大大提高了溴化试剂的利用率,降低了生产成本。
其反应历程如下:
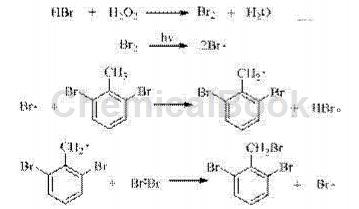
由于Br原子具有很强的吸电子能力,甲基氢的反应活性较低;同时反应的空间位阻较大,使得该反应具有极高的一溴代选择性。
本发明涉及的2,6-二溴溴苄的制备方法,是以2,6-二溴为原料,经溴代反应制得。反应式如下:

其特征在于包括如下步骤:
(1)在有机或无机溶剂存在下,加入化合物2,6-二溴,质量分数为40%的氢溴酸,在光照条件下滴加质量分数为30%的双氧水,反应6~24h;
(2)反应液分别经饱和亚钠溶液、水洗涤、无水钠干燥后减压蒸去溶剂,硅胶柱层析得化合物2,6-二溴溴苄。
主要参考资料
[1] PCT Int. Appl., 2009100169, 13 Aug 2009
[2] CN201110000564.1 一种2,6-二溴溴苄的制备方法


